Protocol
Copyright by Defang Ouyang
Developer: Yunsen Zhang, Chenyu Lin, Zhongmin Zhao, Zheng Wu
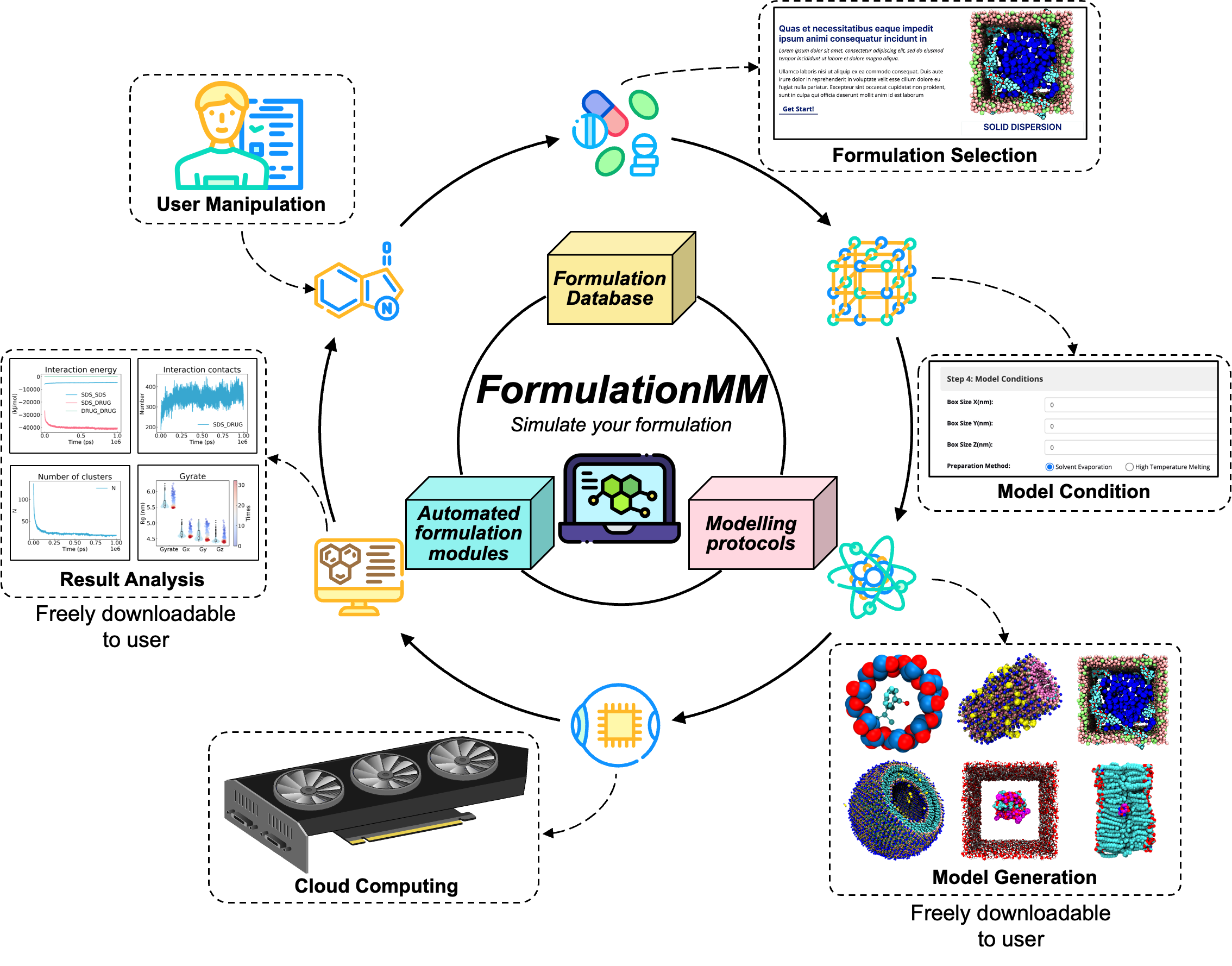
FormulationMM, A state-of-the-art platform, is designed to clarify and predict the atomistic mechanisms of drug formulation based on physics-driven methods. FormulationMM consists of
- Automated formulation software
- Drug formulation database
- Formulation modeling protocols
therefore, supporting streamlined workflow including formulation model generation, molecular dynamics simulation, and results analysis. FormulationMM can automatically generate the forcefield parameter for small drug molecules. FormulationMM supports six prevalent methods of drug formulation, which include
- Cyclodextrin-drug inclusion
- Micelle
- Liposome
- Solid dispersion
- Self-assembling drug nanoparticles
- Drug transmembrane delivery system
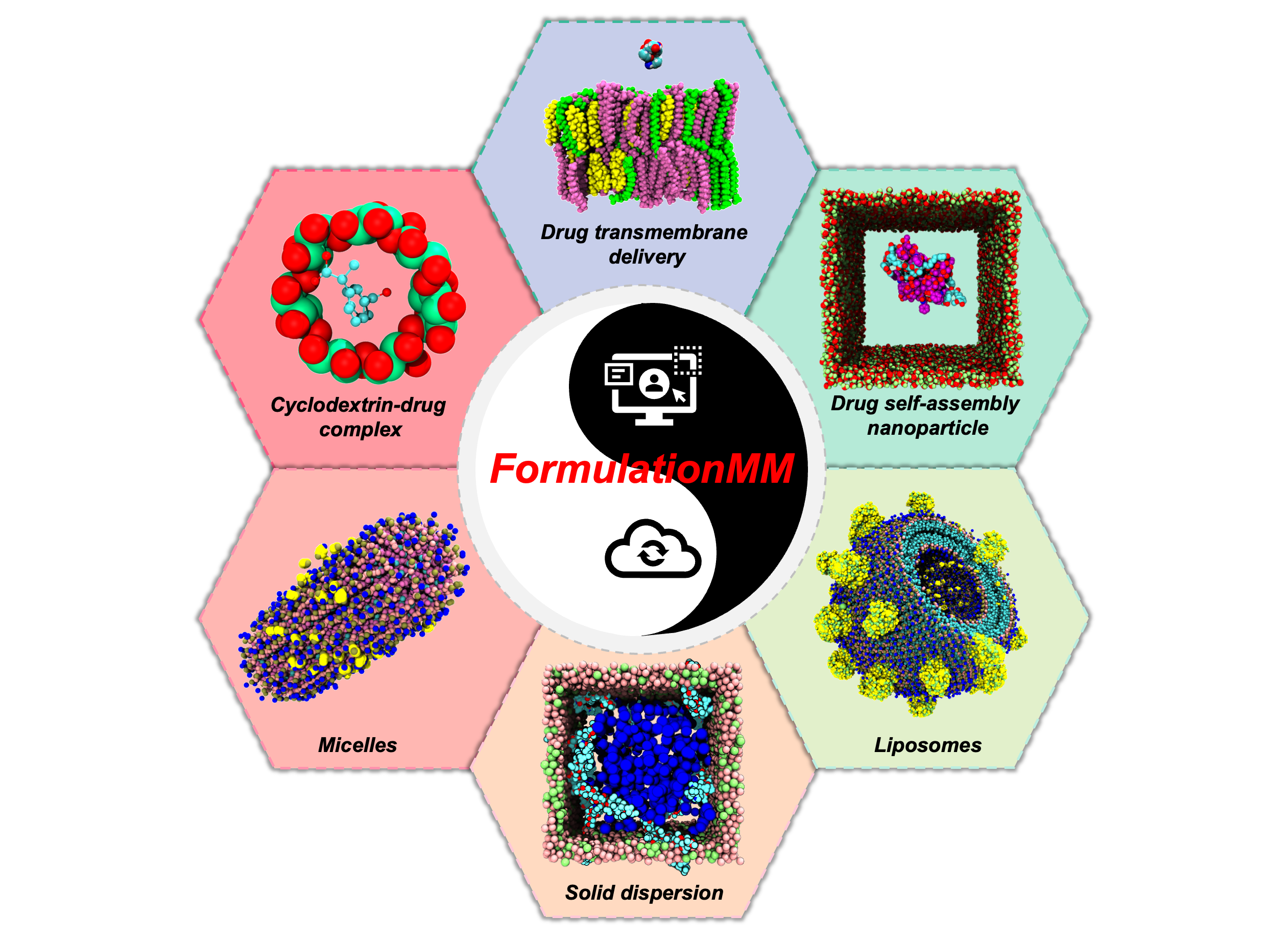
Our computational platform has great potential to revolutionize the research paradigm of drug formulation. FormulationMM is made available through a continuously updated web application (https://formulationmm.computpharm.org).
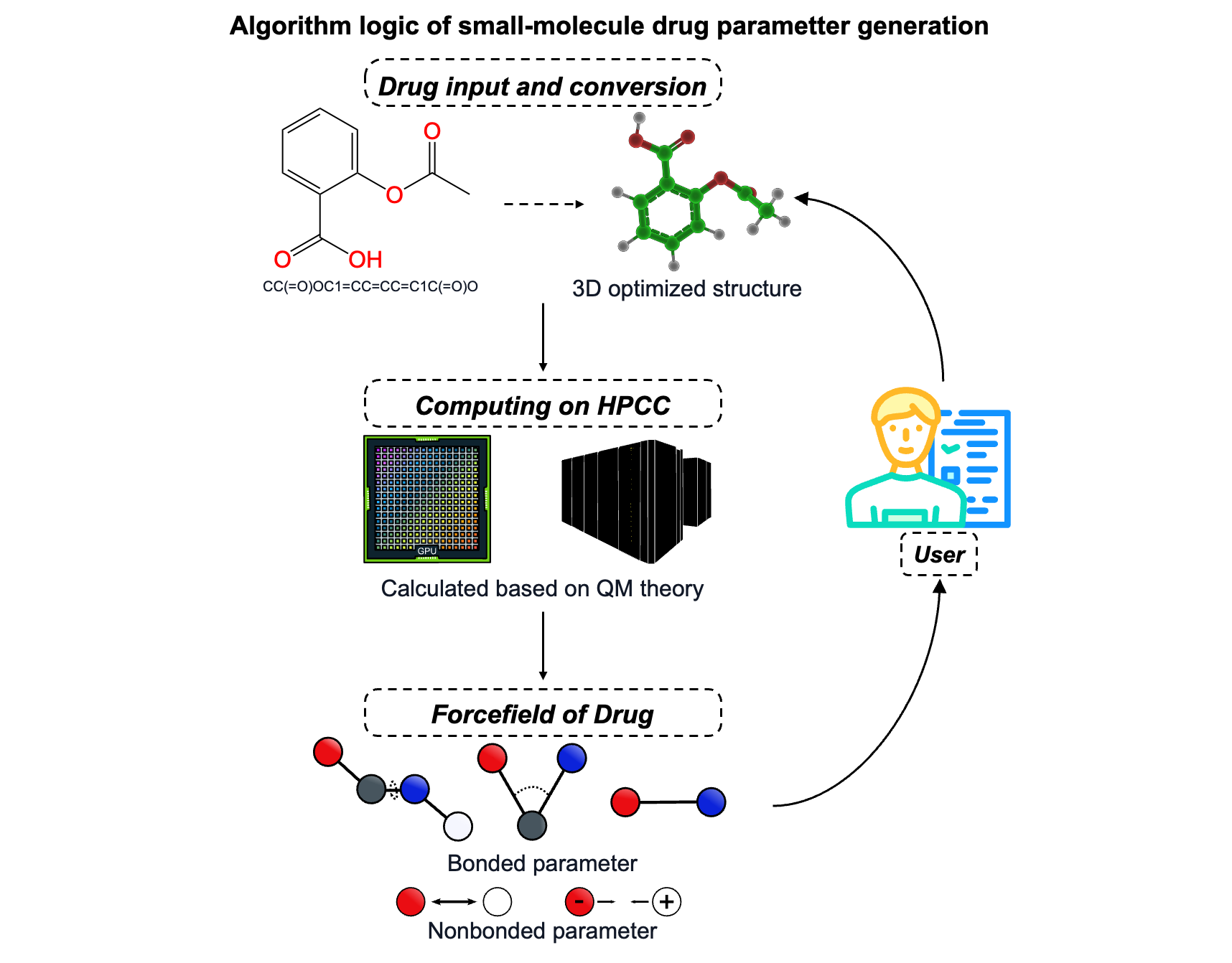
Drug Generation
The central component of the FormulationMM platform, essential for conducting various formulation simulations, begins with the input of molecular structures (See below figure). This information can be provided in various formats, including the Simplified Molecular-Input Line-Entry System (SMILES) or three-dimensional (3D) structures with extensions such as .mol2, .mol, .pdb, .gjf, .xyz, and .cif. Upon receiving the input, the module standardizes molecular representations and uses Open Babel (1) to convert them into .xyz format, which is the required input for subsequent steps. Next, geometry optimization is conducted using the xTB engine with the GFN2-xTB (2) method in implicit water (-g H2O) and --opt extreme settings to ensure accurate minimization. After optimization, the molecule is processed using Multiwfn to extract atomic charges or density-derived information in preparation for quantum chemical calculations. Based on the number of atoms in the system, an appropriate DFT level of theory is automatically selected: small molecules (<30 atoms) are computed using BLYP-D3/def2-TZVP, medium-sized molecules (30–175 atoms) use B97-3c (3,4,5,6), and large molecules (>175 atoms) skip QM and directly proceed to classical parameterization.
For molecules undergoing QM treatment, a full single-point energy calculation is performed using ORCA18. The resulting wavefunction is parsed to compute RESP or AM1-BCC19 charges via Multiwfn (7) and Antechamber (8), depending on the workflow. These charges are integrated into the GAFF2 topology by editing .itp files generated by ACPYPE (9), which wraps around Antechamber and automatically derives force field parameters compatible with GROMACS (10). Finally, the module ensures that residue names are standardized across .itp, .pdb, and .gro files to avoid inconsistencies in downstream molecular dynamics simulation (MD) simulations, enabling automated and reproducible preparation of small molecule topologies for formulation modeling. This integrated workflow seamlessly bridges quantum chemical precision with molecular mechanics compatibility.
This is a tutorial for user to begin with MolPareGen module in FormulationMM platform.
Cyclodextrin
The process begins by accepting user-supplied ligands in formats such as .smi, .mol2, .pdb, and others, which are docked into the cyclodextrin cavity using AutoDock Vina (11). The docking box is automatically defined by computing the cyclodextrin’s center of mass via MDAnalysis (12). The top five poses are extracted and merged with the host structure to construct complete inclusion complexes for simulation. Next, GAFF2-compatible force field parameters are generated for the ligand using the MolParaGen submodule. Cyclodextrin parameters are assumed to be pre-tabulated, and all resulting .itp, .top, and .pdb files are cross-validated and harmonized. The system is then solvated in a cubic box (3×3×3 nm) with SPC/E water (13), and counterions are added to neutralize the system if necessary. Position restraints are applied to both ligand and host to stabilize the system during equilibration.
The user then specifies the cyclodextrin type of interest. Utilizing advanced algorithms, the module identifies the optimal binding centers and prepares the molecules for molecular docking, transforming them into the pdbqt format.
This preprocessing sets the stage for Autodock Vina to generate potential binding poses, serving as a precursor to more in-depth analysis. Acknowledging the constraints of static molecular docking, the module shifts its focus to model the dynamic, water-based environment in which these complexes exist using molecular dynamics simulations.
In this subsequent phase, an animated trajectory of interactions is generated and is a depiction of the drug-cyclodextrin complex under conditions closely resembling physiological states. The final stage of the module focuses on the evaluation of the simulated interactions.
Through the utilization of advanced post-processing tools such as gmx_MMPBSA (14), the module rigorously calculates the MMPB/SA binding free energies. By selecting frames from the balanced segment of the molecular trajectory and incorporating entropy considerations, this stage quantifies the binding affinities, providing a robust measure of the complex's stability.
The calculated binding free energies are not only numerically significant but also align closely with experimental findings, underscoring the predictive accuracy and reliability of our automated tool.
This is a tutorial for user to begin with Cyclodextrin module in FormulationMM platform.
Micelle
The operational framework of the micelle simulation module is designed to provide an in-depth understanding of micelle-mediated drug delivery.
Users initiate the simulation by selecting the surfactant molecules, such as SDS, and their molar ratios, allowing the construction of complex micelle models. The structural data of the drug molecules are input, and the dimensions of the simulation box are set to accommodate the intended micelle size.
The software offers two distinct micelle modeling strategies: 1. Self-assembly Micelle Model: This approach places a specific quantity of drug and surfactant molecules within a predefined box size and solvates them, which is instrumental in studying the micelle formation process and tracking the trajectories of drug molecules during micelle assembly. 2. Preformed Micelle Model: This method constructs pre-assembled micelle models, such as spherical, cylinder-shaped, or capped-cylinder micelles, and introduces a certain number of drug molecules into the system.
This model is used to observe the interaction between a fully formed micelle structure and the drug molecules. Following the construction of the micelle models, the system undergoes molecular dynamics simulations.
The tool automates the analysis of the simulation results, employing measures such as the solvent-accessible surface area to reflect the interactions between the surfactant and the solvent, the radius of gyration to represent the size variations of the micelles and the number density to demonstrate the distribution of drug and surfactant molecules within the micelle. Energy calculations are utilized to reveal the strength of interactions between various components within the micelle and with the encapsulated drug.
Through this sophisticated module, researchers can simulate and analyze the behavior of micelles in drug delivery with exceptional detail.
This is a tutorial for user to begin with Micelle module in FormulationMM platform.
Solid Dispersion
The solid dispersion simulation module offers a structured approach to model the complex physicochemical interactions within solid dispersions.
The workflow is composed of several integral steps that allow for the meticulous construction and analysis of these systems: 1. Carrier and Drug Selection: Users initiate the simulation by selecting the carrier matrix and the drug molecules. Options for carriers include polymers, sugars, or other excipients known to enhance the solubility of APIs. 2. Preparation method: The module allows two different preparation methods: (1) high-temperature melting: we first place the drug molecules into a box, followed by placing the excipients into the same box to generate a simulated initial model.
Next, we perform energy minimization on the system and then subject it to NPT simulations, adjusting the temperature variations to simulate the melting and mixing process of the drug within the excipient matrix under high-temperature experimental conditions. (2) solvent evaporation: we first place the drug and excipients into a container.
Next, we solvate the system, allowing users to choose from various solvents such as ethanol or dichloromethane. Subsequently, we perform energy minimization and NPT equilibrium simulations to ensure thorough dissolution and homogenization of the drug and excipients in the solvent.
Finally, we remove the solvent from the container to simulate the process of solvent evaporation. With the structural model in place, the module conducts molecular dynamics simulations to capture the behavior of the drug within the carrier matrix. The software performs advanced analytical computations to derive meaningful insights. It analyzes key parameters such as the miscibility of the drug and excipient and the amorphous nature of the dispersion.
Through the use of the solid dispersion simulation module, researchers can gain a profound understanding of the drug-carrier interactions, and assess the stability of the dispersion, and predict the dissolution kinetics based on the molecular interactions observed in the simulation.
This is a tutorial for user to begin with Solid Dispersion module in FormulationMM platform.
Liposome
Our tool is designed to streamline the simulation of drug-liposome complexes by integrating model generation, molecular dynamics simulations, and result analysis into a comprehensive system.
The user begins by selecting the constituent phospholipid molecules for the liposome, inputting the structure of the drug molecule of interest, specifying their molar ratios, and choosing a modification approach for the liposome.
The tool offers three options for modification: non-modified, PEG-modified, or protein-modified liposomes. The software then provides two approaches to liposome modeling. The first is a self-assembling liposome model, where a specified number of drug and phospholipid molecules are added to a predefined box size and solvated.
This model can be utilized to study the formation process of liposomes and the trajectories of drug molecules during this process. The second approach involves pre-formed liposome models, where a phospholipid bilayer is first constructed based on the selected lipid types, which then forms the complete vesicle structure into which a specified number of drug molecules are introduced. This is particularly useful for observing the interactions within a fully formed liposome model.
Subsequently, the constructed system undergoes molecular dynamics simulation. The tool automates the analysis of simulation results, employing metrics such as the solvent-accessible surface area of liposomes to reflect the interactions between lipids and the solvent, the radius of gyration to characterize the size changes of liposomes, number density to demonstrate the distribution of drugs and various lipids within the liposome, and energy calculations to indicate the interaction strength between different components of the liposome or with the encapsulated drugs.
Through this sophisticated module, researchers can accurately simulate and analyze the behavior of drug-liposome complexes at an atomic level, gaining insights into the stability, distribution, and interaction mechanisms that underpin their function as drug carriers. This tool significantly enhances the computational efficiency and accuracy, providing a much-needed solution to the challenges inherent in liposome simulation.
This is a tutorial for user to begin with Liposome module in FormulationMM platform.
Drug Nanoparticle
The simulation module for self-assembled nanoparticles is meticulously structured to facilitate the modeling of these complex systems.
The process flow is designed to reflect the multifaceted nature of nanoparticle formation and drug encapsulation:
- Molecule Selection and Configuration: The initial step involves selecting the molecular components that will form the nanoparticles. For paclitaxel and curcumin nanoparticles, users can choose from a variety of surfactants and polymers that facilitate self-assembly.
- Nanoparticle Assembly Modeling: The module then allows for the design of the nanoparticle structure, incorporating the drug molecules into the assembly matrix.
- Solvent and Environmental Conditions: The simulation considers the solvent and environmental parameters crucial for nanoparticle stability and drug release. It emulates physiological conditions to predict the behavior of the nanoparticles upon administration.
- Dynamics Simulation and Result Analysis: With the structural model established, the module conducts molecular dynamics simulations to observe the self-assembly process and the resulting nanoparticle structure. It evaluates the encapsulation efficiency, stability of the drug within the nanoparticle, and the potential release profile.
The self-assembled nanoparticle simulation module provides powerful software for researchers to design and optimize nanoparticle formulations for drugs like paclitaxel and curcumin. By facilitating a deep understanding of the self-assembly process and the resulting nanoparticle characteristics, the module aids in the refinement of drug delivery strategies.
This is a tutorial for user to begin with Self-assembly drug nanoparticle module in FormulationMM platform.
Drug Transmembrane
This module provides an immersive simulation experience. Users begin by selecting appropriate lipid molecules to construct a biological membrane model that could vary from a simple plasmalemma to complex endoplasmic membranes.
They then input the drug molecule's structural data and establish the dimensions of the simulation box, tailoring it to the scale and scope of their study. Once the structural setup is complete, the tool autonomously adjusts the box size, sets up the initial position of the drug molecule at an appropriate distance from the membrane surface, and embarks on a detailed equilibration protocol.
This includes a six-step equilibration, embracing energy minimization to remove any steric clashes or inappropriate geometries, followed by NVT and NPT ensemble simulations to acclimate the system to the desired temperature and pressure while maintaining the integrity of the initial conditions through careful application of positional restraints. Upon achieving a stable system, the tool transitions to the umbrella sampling simulation phase.
- In the first part of this phase, umbrella pulling is performed, where the tool applies a calibrated force to shepherd the drug molecule across the cell membrane at a controlled rate and trajectory, acquiring a comprehensive set of enhanced sampling trajectories.
- The second part involves meticulous window extraction and sampling, culminating in the construction of a definitive PMF curve. Researchers can leverage this curve to quantitatively assess the transmembrane ability of the drug molecule, effectively mapping out the energy profile of the permeation pathway.
This module, with its state-of-the-art simulation capabilities, affords researchers an unparalleled level of detail in understanding the transmembrane transport of drugs. By dissecting the energetic nuances of drug-membrane interactions, the tool contributes substantially to the predictive modeling of drug absorption profiles.
This is a tutorial for user to begin with Drug transmembrane module in FormulationMM platform.
Reference
(1) O'Boyle, N.M. et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics 3, 33 (2011).
(2) Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. Journal of Chemical Theory and Computation 15, 1652-1671 (2019).
(3) Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. The Journal of Chemical Physics 132 (2010).
(4) Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. Journal of Computational Chemistry 32, 1456-1465 (2011).
(5) Brandenburg, J.G., Bannwarth, C., Hansen, A. & Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. The Journal of Chemical Physics 148 (2018).
(6) Brandenburg, J.G., Bates, J.E., Sun, J. & Perdew, J.P. Benchmark tests of a strongly constrained semilocal functional with a long-range dispersion correction. Physical Review B 94, 115144 (2016).
(7) Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. Journal of Computational Chemistry 33, 580-592 (2012).
(8) Salomon-Ferrer, R., Case, D.A. & Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Computational Molecular Science 3, 198-210 (2013).
(9) Sousa da Silva, A.W. & Vranken, W.F. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Research Notes 5, 367 (2012).
(10) Van Der Spoel, D. et al. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry 26, 1701-1718 (2005).
(11) Trott, O. & Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31, 455-461 (2010).
(12) Michaud-Agrawal, N., Denning, E.J., Woolf, T.B. & Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. Journal of Computational Chemistry 32, 2319-2327 (2011).
(13) Mark, P. & Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. The Journal of Physical Chemistry A 105, 9954-9960 (2001).
(14) Valdés-Tresanco, M.S., Valdés-Tresanco, M.E., Valiente, P.A. & Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. Journal of Chemical Theory and Computation 17, 6281-6291 (2021).